Introduction
Integrons are major genetic element, notorious for their major implication in the spread of antibiotic resistance genes. More generally, integrons are gene-capturing platform, whose broader evolutionary role remains poorly understood. IntegronFinder is able to detect with high accuracy integron in DNA sequences. It is accurate because it combines the use of HMM profiles for the detection of the essential protein, the site-specific integron integrase, and the use of Covariance Models for the detection of the recombination site, the attC site.
How does it work ?
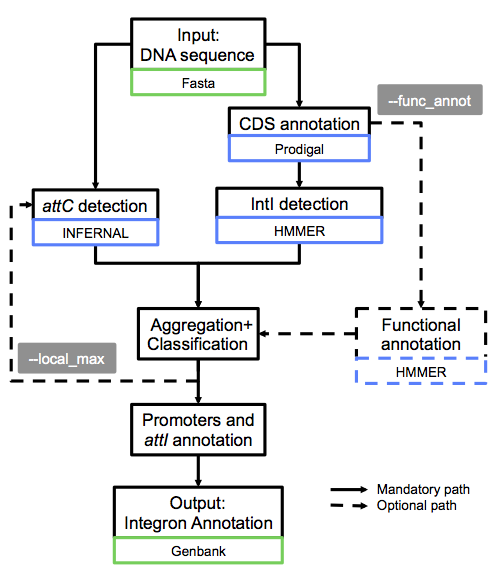
For each sequence in the input file:
First, IntegronFinder annotates the DNA sequence’s CDS with Prodigal.
Second, IntegronFinder detects independently integron integrase and attC recombination sites. The Integron integrase is detected by using the intersection of two HMM profiles:
one specific of tyrosine-recombinase (PF00589)
one specific of the integron integrase, near the patch III domain of tyrosine recombinases.
The attC recombination site is detected with a covariance model (CM), which models the secondary structure in addition to the few conserved sequence positions.
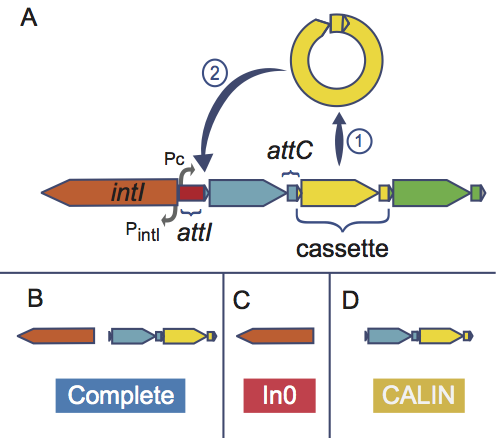
Third, the results are integrated, and IntegronFinder distinguishes 3 types of elements:
- complete integron (panel B above)
Integron with integron integrase nearby attC site(s)
- In0 element (panel C above)
Integron integrase only, without any attC site nearby
- CALIN element (panel D above)
Cluster of attC sites Lacking INtegrase nearby. A rule of thumb to avoid false positive is to filter out singleton of attC site.
IntegronFinder can also annotate gene cassettes (CDS nearby attC sites) using AMRFinderPlus, a database of HMM profiles aiming at annotating antibiotic resistance genes. This database is provided but the user can add any other HMM profiles database of its own interest.
When available, IntegronFinder annotates the promoters and attI sites by pattern matching.
Does it work ?
Yes! The estimated sensitivity is 61% on average with the default option and goes up to 88% with the --local_max option.
The missing attC sites are usually at the end of the array. The False positive rate with the --local_max
option is estimated between 0.03 False Positive per Megabases (FP/Mb) to 0.72 FP/Mb. This leads to a probability of
finding 2 consecutive false attC sites within 4kb between 4.10^-6 and 7.10^-9. Overall, the probability of finding
an integron in a chromosome (including finding a part of it) is more than 95%. Finally, these parameters
do not depend on the G+C percent of the given replicon. See the paper for more information (freely accessible).
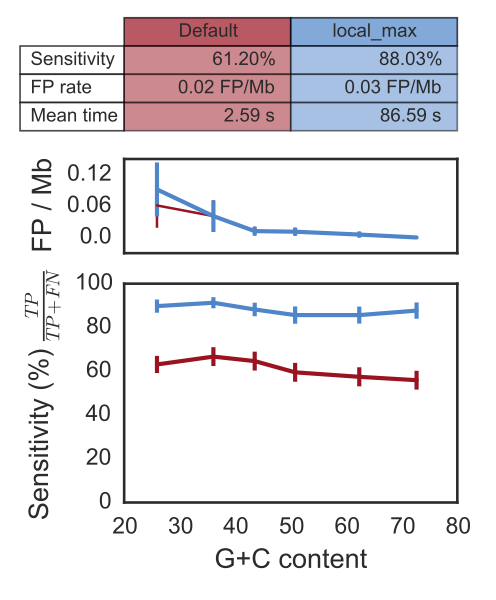
The time in the table correspond to the average time per run with a pseudogenome having attC sites on a Mac Pro, 2 x 2.4 GHz 6-Core Intel Xeon, 16 Gb RAM, with options –cpu 20 and –no-proteins.
Note
The time does not vary depending of the mode (default or local_max), and is about a couple of second, if the replicon does not contain any attC site.
