Quick start
We assume here that the program is installed.
You can see all available options with:
integron_finder -h
For impatient
Go to the directory containing your input file(s), or specify the path to that file and call:
integron_finder mysequences.fst
or:
integron_finder path/to/mysequences.fst
It will perform a search, and outputs the results in a directory called
Results_Integron_Finder_mysequences.
Input and Outputs
Inputs
integron_finder can take as an input:
a fasta file
a multi-fasta file
many (multi-)fasta files
several replicon in gembase format
Gembase format
Integron_Finder can use gembase formatted protein files instead of re-annotating genes with Prodigal. This feature enables the user to use Integron_Finder with its own annotations. The gembase format is the typical result of PanACoTA’s annotation step
Integron_Finder support gembase format v1 and v2
Gembase v1
It must contain, at least, a LSTINF and a Protein file per replicon. Folder structure must have the following architecture.
gembase/
├── Genes
│ ├── ACBA.0917.00019.gen # genome in fasta format one seq/gene
│ └── ESCO001.C.00001.C001.gen
├── LSTINFO
│ ├── ACBA.0917.00019.lst # information in space separated values
│ └── ESCO001.C.00001.C001.lst
├── Proteins
│ ├── ACBA.0917.00019.prt # proteins in fasta format
│ └── ESCO001.C.00001.C001.prt
└── Replicons
├── ACBA.0917.00019.fna # genome in fasta format one seq/replicon
└── ESCO001.C.00001.C001.fst
5 first sequences headers in gembase/Genes/ACBA.0917.00019.gen file
>ACBA.0917.00019.b0001_00001 1215 tyrS | Tyrosine--tRNA ligase | 6.1.1.1 | similar to AA sequence:UniProtKB:P41256
>ACBA.0917.00019.i0001_00002 1128 anmK | Anhydro-N-acetylmuramic acid kinase | 2.7.1.170 | similar to AA sequence:UniProtKB:Q8EHB5
>ACBA.0917.00019.i0001_00003 846 ephA | Epoxide hydrolase A | 3.3.2.10 | similar to AA sequence:UniProtKB:I6YGS0
>ACBA.0917.00019.i0001_00004 336 erpA | Iron-sulfur cluster insertion protein ErpA | NA | similar to AA sequence:UniProtKB:P45344
>ACBA.0917.00019.i0001_00005 1005 NA | hypothetical protein | NA | NA
...
5 first sequences headers in gembase/Genes/ACBA.0917.00019.gen file
>ACBA.0917.00019.b0001_00001 1215 tyrS | Tyrosine--tRNA ligase | 6.1.1.1 | similar to AA sequence:UniProtKB:P41256
>ACBA.0917.00019.i0001_00002 1128 anmK | Anhydro-N-acetylmuramic acid kinase | 2.7.1.170 | similar to AA sequence:UniProtKB:Q8EHB5
>ACBA.0917.00019.i0001_00003 846 ephA | Epoxide hydrolase A | 3.3.2.10 | similar to AA sequence:UniProtKB:I6YGS0
>ACBA.0917.00019.i0001_00004 336 erpA | Iron-sulfur cluster insertion protein ErpA | NA | similar to AA sequence:UniProtKB:P45344
>ACBA.0917.00019.i0001_00005 1005 NA | hypothetical protein | NA | NA
...
5 first lines in gembase/LSTINFO/ACBA.0917.00019.lst
266 1480 C CDS ACBA.0917.00019.b0001_00001 tyrS | Tyrosine--tRNA ligase | 6.1.1.1 | similar to AA sequence:UniProtKB:P41256
1560 2687 D CDS ACBA.0917.00019.i0001_00002 anmK | Anhydro-N-acetylmuramic acid kinase | 2.7.1.170 | similar to AA sequence:UniProtKB:Q8EHB5
2815 3660 D CDS ACBA.0917.00019.i0001_00003 ephA | Epoxide hydrolase A | 3.3.2.10 | similar to AA sequence:UniProtKB:I6YGS0
3716 4051 C CDS ACBA.0917.00019.i0001_00004 erpA | Iron-sulfur cluster insertion protein ErpA | NA | similar to AA sequence:UniProtKB:P45344
4176 5180 C CDS ACBA.0917.00019.i0001_00005 NA | hypothetical protein | NA | NA
...
all sequences headers in gembase/Replicons/ACBA.0917.00019.fna
>ACBA.0917.00019.0001
>ACBA.0917.00019.0002
Integron_finder will use LSTINF and Proteins folders. Hence if you want to analyze a replicon located in a gembase, the command line should look like
integron_finder –gembase data/Replicons/ACBA.0917.00019.fna
Gembase v2
In V2 format, there is one file per genome. If there are several chromosomes or chromosomes plus plasmids, phages, … all the sequences are in the same file.
Example of Gembase v2 architecture:
Gembase_v2_extraction/
├── Genes
│ ├── VICH001.0523.00090.gen
│ ├── VICH001.0523.00091.gen
│ ├── ...
├── LST
│ ├── VICH001.0523.00090.lst
│ ├── VICH001.0523.00091.lst
│ ├── ...
├── Microbial0523-genomes.info
├── Microbial0523-replicons.info
├── Proteins
│ ├── VICH001.0523.00090.prt
│ ├── VICH001.0523.00091.prt
│ ├── ...
├── RNA
│ ├── VICH001.0523.00090.rna
│ ├── VICH001.0523.00091.rna
│ ├── ...
├── Replicons
│ ├── VICH001.0523.00090.fna
│ ├── VICH001.0523.00091.fna
│ ├── ...
└── gff
├── VICH001.0523.00090.gff3
├── VICH001.0523.00091.gff3
├── ...
5 first sequences headers in gembase/Genes/VICH001.0523.00090.gen file
>VICH001.0523.00090.001C_00001 C ATG TAA 1 2961008 1128 mltC FKV26_RS00005 WP_001230176.1 membrane-bound_lytic_murein_transglycosylase_MltC
ATGAGAAAGTTAGTGTTATGTATCACCGCTCTATTATTGAGTGGCTGTAGTCGTGAATTTATCGAGAAAATTTACGATGTTGATTACGAGCCCACTAACCGTTTCGCCAAGAACTTAGCCGAATTACCCGGACAGTTTCAGAAAGACACCGCCGCACTGGATGCATTAATCAACAGCTTCTCCGGCAATATCGAAAAACGTTGGGGGCGCCGCGAGCTA
>VICH001.0523.00090.001C_00002 C ATG TAG 913 1185 273 NA FKV26_RS00010 WP_000124739.1 oxidative_damage_protection_protein
ATGGCTCGCACCGTATTTTGTACCCGATTGCAGAAAGAAGCCGATGGCTTGGATTTCCAACTCTATCCCGGAGAACTGGGCAAACGCATTTTCGACAACATCTGCAAAGAAGCTTGGGCACAATGGCAGACCAAACAGACCATGCTGATCAATGAAAAAAAACTCAACATGATGGATCCTGAGCACCGCAAATTGCTGGAGCAAGAAATGGTGAGCTTC
>VICH001.0523.00090.001C_00003 C GTG TAA 1199 2260 1062 mutY FKV26_RS00015 WP_001995048.1 A/G-specific_adenine_glycosylase
...
VICH001.0523.00090 contains 2 chromosomes 001C and 002C
5 first lines of chromosome 1 and 2 in gembase/LST/VICH001.0523.00090.lst
1 2961008 C CDS VICH001.0523.00090.001C_00001 FKV26_RS00005 mltC WP_001230176.1 membrane-bound_lytic_murein_transglycosylase_MltC 4.2.2.- COORDINATES:_similar_to_AA_sequence:RefSeq:WP_001230183.1 GO:0008933_-_lytic_transglycosylase_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
913 1185 C CDS VICH001.0523.00090.001C_00002 FKV26_RS00010 NA WP_000124739.1 oxidative_damage_protection_protein NA COORDINATES:_similar_to_AA_sequence:RefSeq:WP_005482430.1 GO:0005506_-_iron_ion_binding_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
1199 2260 C CDS VICH001.0523.00090.001C_00003 FKV26_RS00015 mutY WP_001995048.1 A/G-specific_adenine_glycosylase 3.2.2.31 COORDINATES:_similar_to_AA_sequence:RefSeq:WP_014505785.1 GO:0019104_-_DNA_N-glycosylase_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
2500 3219 D CDS VICH001.0523.00090.001C_00004 FKV26_RS00020 trmB WP_000005581.1 tRNA_(guanosine(46)-N7)-methyltransferase_TrmB 2.1.1.33 COORDINATES:_similar_to_AA_sequence:RefSeq:WP_001898008.1 GO:0008176_-_tRNA_(guanine-N7-)-methyltransferase_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
3330 4250 D CDS VICH001.0523.00090.001C_00005 FKV26_RS00025 glsB WP_000805054.1 glutaminase_B 3.5.1.2 COORDINATES:_similar_to_AA_sequence:RefSeq:WP_002046360.1 GO:0004359_-_glutaminase_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
...
15 1373 D CDS VICH001.0523.00090.002C_02705 FKV26_RS13475 glpT WP_000462380.1 glycerol-3-phosphate_transporter NA COORDINATES:_similar_to_AA_sequence:RefSeq:WP_019829515.1 GO:0015169_-_glycerol-3-phosphate_transmembrane_transporter_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
1527 2594 D CDS VICH001.0523.00090.002C_02706 FKV26_RS13480 glpQ WP_000917229.1 glycerophosphodiester_phosphodiesterase 3.1.4.46 COORDINATES:_similar_to_AA_sequence:RefSeq:WP_005524061.1 GO:0008081_-_phosphoric_diester_hydrolase_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
2647 3327 C CDS VICH001.0523.00090.002C_02707 FKV26_RS13485 NA WP_001177500.1 Crp/Fnr_family_transcriptional_regulator NA COORDINATES:_similar_to_AA_sequence:RefSeq:WP_001887744.1 NA Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
3467 4198 C CDS VICH001.0523.00090.002C_02708 FKV26_RS13490 NA WP_000027384.1 nucleoside_phosphorylase NA COORDINATES:_similar_to_AA_sequence:RefSeq:WP_001897923.1 NA Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
4195 4920 C CDS VICH001.0523.00090.002C_02709 FKV26_RS13495 pnuC WP_001995272.1 nicotinamide_riboside_transporter_PnuC NA COORDINATES:_similar_to_AA_sequence:RefSeq:WP_001933049.1 GO:0015663_-_nicotinamide_mononucleotide_transmembrane_transporter_activity_[Evidence_IEA] Derived_by_automated_computational_analysis_using_gene_prediction_method:_Protein_Homology.
5 sequences of chromosome 1 and 2 in gembase/Proteins/VICH001.0523.00090.prt
>VICH001.0523.00090.001C_00001 C ATG TAA 1 2961008 375 mltC FKV26_RS00005 WP_001230176.1 membrane-bound_lytic_murein_transglycosylase_MltC
MRKLVLCITALLLSGCSREFIEKIYDVDYEPTNRFAKNLAELPGQFQKDTAALDALINSFSGNIEKRWGRRELKIAGKNNYVKYIDNYLSRSEVNFTEGRIIVETVSPIDPKAHLRNAIITTLLTPDDPAHVDLFSSKDIELKGQPFLYQQVLDQDGQPIQWSWRANRYADY
>VICH001.0523.00090.001C_00002 C ATG TAG 913 1185 90 NA FKV26_RS00010 WP_000124739.1 oxidative_damage_protection_protein
MARTVFCTRLQKEADGLDFQLYPGELGKRIFDNICKEAWAQWQTKQTMLINEKKLNMMDPEHRKLLEQEMVSFLFEGKEVHIEGYTPPAK
...
>VICH001.0523.00090.002C_02705 D ATG TAA 15 1373 452 glpT FKV26_RS13475 WP_000462380.1 glycerol-3-phosphate_transporter
MFELFKPTAHTQRLPSDKVDSVYSRLRWQLFIGIFVGYAGYYLVRKNFSLAMPYLIEQGFSRGDLGVALGAVSIAYGLSKFLMGNVSDRSNPRYFLSAGLLLSALVMFCFGFMPWATGSITAMFILLFLNGWFQGMGWPACGRTMVHWWSRKER
>VICH001.0523.00090.002C_02706 D ATG TAA 1527 2594 355 glpQ FKV26_RS13480 WP_000917229.1 glycerophosphodiester_phosphodiesterase
MLKPFSLSLLALACSTSLFSSIVSAEPIVIAHRGASGYLPEHTLEAKTLAYAMKPDYIEQDVVMTKDDQLVVLHDHYLDRVTDVAERFPNRARADGRYYAIDFTLAEIKTLRVTEGFNIDDQGKKVAGFPDRFPIWKGDFTVPTLAEEIELIQGLNKTLG
all sequences headers in gembase/Replicons/VICH001.0523.00090.fna
>VICH001.0523.00090.001C 2023-05-16 2961008 bp Vibrio cholerae O1 strain AAS91 chromosome 1, complete sequence
>VICH001.0523.00090.002C 2023-05-16 1124701 bp Vibrio cholerae O1 strain AAS91 chromosome 2, complete sequence
Integron_finder will use LST (or LSTINF(O) for Gembase v1) and Proteins folders. Hence if you want to analyze a replicon located in a gembase, the command line should look like
integron_finder --gembase data/My_base/Replicons/ACBA.0917.00019.fna
Note
If the replicon you want to analyze is not in the gembase directory, but you still want to take advantage of the gembase annotation, then you have to specify the –gembase-path option to indicate where to find it. A typical command line could be:
integron_finder --gembase --gembase-path data/gembase/ My_replicons/ACBA.0917.00019.fna
Note
The gembase format implies different default topology (Topology)
Custom protein file
Integron_Finder allow the user to provide it’s own protein file with it’s own annotations instead of using prodigal (default). In this case you have to specified the protein file to use with the option –prot-file and the path to the parser to extract information from this file –annot-parser. A typical command line could be:
integron_finder –prot-file <path/to/custom/protein /file> –annot-parser <my_annot_paser.py> <replicon_path>
The annotation parser must be a python file (with the .py extension’) with one function called description_parser This function must take one argument which is the header of a fasta sequence header (without the first character >) and must return a tuple with 4 elements:
sequence id: the id of the sequence (string)
start: the beginning position of the protein on the genome (positive int)
stop: the end position of the protein on the genome (positive int)
strand: the strand 1 if the protein os coded on the direct strand or -1 on the reverse
Below a description_parser to parse annotation in prodigal format:
from typing import Tuple def description_parser(seq_header: str) -> Tuple[int, int, int, int]: """ Extract description features from protein sequence fasta header :param str seq_header: the fasta header of a sequence (without '>' char) :return: features - sequence id (string) - start the beginning of the protein (positive integer) - stop the end position of the protein (positive integer) - strand 1 if prot is coded on direct strand or -1 if it's on reverse :rtypes: tuple (str, int, int, 1/-1) """ id_, start, stop, strand, *_ = seq_header.split(" # ") start = int(start) stop = int(stop) strand = int(strand) return id_, start, stop, strand
Outputs
By default, integron_finder will output 3 files under Results_Integron_Finder_mysequences:
mysequences.integrons: A file with all integrons and their elements detected in all sequences in the input file.mysequences.summary: A summary file with the number and type of integrons per sequence.integron_finder.out: A copy standard output. The stdout can be silenced with the argument--mute
The amount of log in the standard output can be controlled with --verbose for more or --quiet for less,
and both are cumulative arguments, eg. -vv or -qq.
Other files can be created on demand:
--gbk: Creates a Genbank files with all the annotations found (present in the.integronsfile)--pdf: Creates a simple pdf graphic with complete integrons--split-results: Creates a.integronsa.summaryfile per replicon if the input is a multifasta file.--keep-tmp: Keep temporary files. See Keep intermediate files for more.
For everyone
Note
The different options will be shown separately, but they can be used altogether unless otherwise stated.
Thorough local detection
This option allows a much more sensitive search of attC sites. It will be slower if integrons are found, but will be as fast if nothing is detected.
integron_finder mysequences.fst --local-max
CALIN detection
By default CALIN are reported if they are composed of at least 2 attC sites, in order to avoid false positives. This value was chosen as CALIN with 2 attC sites were unlikely to be false positive. The probability of a false CALIN with at least 2 attC sites within 4kb was estimated between 4.10^-6 and 7.10^-9. However, one can modify this value with the option –calin-threshold and use a lower or higher value depending on the risk one is willing to take:
integron_finder mysequences.fst --calin-threshold 1
Note
If --local-max is called, it will run around CALINs with single attC sites, even if --calin-threshold is 2.
The filtering step is done after the search with local max in that case.
Functional annotation
This option allows to annotate cassettes given HMM profiles. As AMRFinderPlus database is distributed, to annotate antibiotic resistance genes, just use:
integron_finder mysequences.fst --func-annot
IntegronFinder will look in the directory
Integron_Finder-x.x/data/Functional_annotation and use all .hmm files
available to annotate. By default, there is only NCBIfam-AMRFinder.hmm, but one can
add any other HMM file here. Alternatively, if one wants to use a database which
is present elsewhere on the user’s computer without copying it into that
directory, one can specify the following option
integron_finder mysequences.fst --path_func_annot bank_hmm
where bank_hmm is a file containing one absolute path to a hmm file per
line, and you can comment out a line
~/Downloads/Integron_Finder-x.x/data/Functional_annotation/NCBIfam-AMRFinder.hmm
~/Documents/Data/Pfam-A.hmm
# ~/Documents/Data/Pfam-B.hmm
Here, annotation will be made using Pfam-A et NCBIfam-AMRFinder, but not Pfam-B. If a protein is hit by 2 different profiles, the one with the best e-value will be kept.
Search for promoter and attI sites
By default integron_finder look for attC sites and site-specific integron integrase,,
If you want to search for known promoters (integrase, Pc-int1 and Pc-int3) and AttI sites
in integrons elements you need to add the --promoter-attI option on the command line.
Keep intermediate results
Integrons finder needs some intermediate results to run completely.
It includes notably the protein file in fasta (mysequences.prt), but also the outputs from hmmer and infernal.
A folder containing these outputs is generated for each replicon and have name tmp_<replicon_id>
This directory is removed at the end. You can keep this directory to analyse further each integron_finder steps
with the option --keep-tmp. Using this argument allows you to rerun integron_finder
on the same sequences without redetecting proteins and attC sites. It is useful if one wants to change
clustering parameters, evalues of attC sites, or size of them. Note that it won’t search for new attC sites
so it is better to start with relaxed parameters and then rerun integron_finder with more strict parameters.
See the section for integron diggers for more informations
For each tmp file, there are:
<replicon_id>.fst: a single fasta file with the replicon_name<replicon_id>.prt: a multifasta file with the sequences of the detected proteins.<replicon_id>_intI_table.res: hmm result for the intI hmm profile in tabular format<replicon_id>_intI.res: hmm result for the intI hmm profile<replicon_id>_phage_int_table.res: hmm result for the tyrosine recombinase hmm profile in tabular format<replicon_id>_phage_int.res: hmm result for the tyrosine recombinase hmm profile in tabular format<replicon_id>_attc_table.res: cmsearch result for the attC sites covariance model in tabular format<replicon_id>_attc.res: significant (according toevalue-attc) attC sites aligned in stockholm formatintegron_max.pickle: pickle file sointegron_finderreuse this instead of re-running the local_max part
Topology
For regular sequence file format
By default, IntegronFinder assumes that
your replicon is considered as circular if there is only one replicon in the input file.
your replicons are considered as linear if there are several replicons in the input file.
For sequence in GemBase format
By default, IntegronFinder assumes that
Your replicon is considered as circular if it’s a Complete Relicon and it’s a Chromosome or Plasmid. Even there are several replicons in the same file.
If your replicons are a Phage, Other or a Draft. They will be considered by default as linear.
Whatever The sequence format
However, you can change this default behavior and specify the default topology with options
--circ or --lin on the command line:
integron_finder --lin mylinearsequence.fst
integron_finder --circ mycircularsequence.fst
If you have multiple replicon in the input file with different topologies you can specify a topology for each replicon by providing a topology file. The syntax for the topology file is simple:
one topology by line
one line start by the seqid followed by ‘circ’ or ‘lin’ for circular or linear topologies.
example:
seq_id_1 circ
seq_id_2 lin
You can also mix the options --circ or --lin with option --topology-file:
integron_finder --circ --topology-file path/to/topofile mysequencess.fst
In the example above the default topology is set to circular. The replicons specified in topofile supersede the default topology.
Warning
However, if the replicon is smaller than 4 x dt
(where dt is the distance threshold, so 4kb by default), the replicon is considered linear
to avoid clustering problem.
The topology used to searching integron is report in the *.integrons file
For big data people
Parallelization
The time limiting part are HMMER (search integrase) and INFERNAL (search attC sites). So if you have to analyze one or few replicons the user can set the number of CPU used by HMMER and INFERNAL:
integron_finder mysequences.fst --cpu 4
Default is 1.
Warning
Increasing too much (usually above 4) the number of CPUs used may lower the performance of the software. Please refer to the documentation of HMMER and INFERNAL for more details.
If you want to deal with a fasta file with a lot of replicons (from 10 to more than thousand) we provide a workflow to parallelize the execution of the data. This mean that we cut the data input into chunks (by default of one replicon) then execute IntegronFinder in parallel on each replicon (the number of parallel tasks can be limited) then aggregate the results in one global summary. The workflow use the nextflow framework and can be run on a single machine or a cluster.
First, you have to install nextflow first, and integron_finder. Then we provide 2 files (you need to download them from the IntegronFinder github repo.)
parallel_integron_finder.nf which is the workflow itself in nextflow syntax
nextflow.config which is a configuration file to execute the workflow.
The workflow file should not be modified. Whereas the profile must be adapted to the local architecture.
- The file nextflow.config provide for profiles:
a standard profile for local use
a cluster profile
a standard profile using apptainer container
a cluster profile using apptainer container
so now install nextflow. If you have capsule error like
CAPSULE EXCEPTION: Error resolving dependencies. while processing attribute Allow-Snapshots: false (for stack trace, run with -Dcapsule.log=verbose) Unable to initialize nextflow environment
install nextflow (>=0.29.0) as follow (change the nextflow version with the last release)
wget -O nextflow http://www.nextflow.io/releases/v0.30.2/nextflow-0.30.2-all chmod 777 nextflow
for more details see: https://github.com/nextflow-io/nextflow/issues/770#issuecomment-400384617
How to get parallel_integron_finder
The release contains the workflow parallel_integron_finder.nf and the nextflow.config at the top level of the archive But If you use pip to install Integron_Finder you have not easily access to them. But they can be downloaded or executed directly by using nextflow.
to download it
nextflow pull gem-pasteur/Integron_Finder
to get the latest version or use -r option to specify a version
nextflow pull -r release_2.0 gem-pasteur/Integron_Finder
to see what you download
nextflow see Integron_Finder
to execute it directly
nextflow run gem-pasteur/Integron_Finder -profile standard --replicons all_coli.fst --circ
or:
nextflow run -r release_2.0 gem-pasteur/Integron_Finder -profile standard --replicons all_coli.fst --circ
standard profile
This profile is used if you want to parallelize IntegronFinder on your machine. You can specify the number of tasks in parallel by setting the queueSize value
standard {
executor {
name = 'local'
queueSize = 7
}
process{
executor = 'local'
$integron_finder{
errorStrategy = 'ignore'
cpu=params.cpu
}
}
}
If you installed IntegronFinder with apptainer, just uncomment the container line in the script, and set the proper path to the container.
All options available in non parallel version are also available for the parallel one.
except the --outdir which is not available and --replicons option which is specific to the parallelized version.
--replicons allows to specify the path of a file containing the replicons.
A typical command line will be:
./parallel_integron_finder.nf -profile standard --replicons all_coli.fst --circ
Note
Joker as * or ? can be used in path to specify several files as input.
But do not forget to protect the wild card from the shell for instance by enclosing your glob pattern with simple quote.
nextflow run -profile standard parallel_integron_finder.nf --replicons 'replicons_dir/*.fst'
Two asterisks, i.e. **, works like * but crosses directory boundaries.
Curly brackets specify a collection of sub-patterns.
nextflow run -profile standard parallel_integron_finder.nf --replicons 'data/**.fa'
nextflow run -profile standard parallel_integron_finder.nf --replicons 'data/**/*.fa'
nextflow run -profile standard parallel_integron_finder.nf --replicons 'data/file_{1,2}.fa'
The first line will match files ending with the suffix .fa in the data folder and recursively in all its sub-folders. While the second one only match the files which have the same suffix in any sub-folder in the data path. Finally the last example capture two files: data/file_1.fa, data/file_2.fa
More than one path or glob pattern can be specified in one time using comma. Do not insert spaces surrounding the comma
nextflow run -profile standard parallel_integron_finder --replicons 'some/path/*.fa,other/path/*.fst'
The command above will analyze all files ending by .fa in /some/path with .fst extension in other/path
For further details see: https://www.nextflow.io/docs/latest/channel.html#frompath
Note
The option –outdir is not allowed. Because you can specify several replicon files as input, So in this circumstances specify only one name for the output is a none sense.
Note
The options starting with one dash are for nextflow workflow engine, whereas the options starting by two dashes are for integron_finder workflow.
Note
Replicons will be considered linear by default (see above), here we use –circ to consider replicons circular.
Note
If you specify several input files, the split and merge steps will be parallelized.
If you execute this line, 2 kinds of directories will be created.
One named work containing lot of subdirectories this for all jobs launch by nextflow.
Directories named Results_Integron_Finder_XXX where XXX is the name of the replicon file. So, one directory per replicon file will be created. These directories contain the final results as in non parallel version.
cluster profile
The cluster profile is intended to work on a cluster managed by SLURM. If your cluster is managed by an other drm replace executor name by the right value (see nextflow supported cluster )
You can also manage
The number of tasks in parallel with the executor.queueSize parameter (here 500). If you remove this line, the system will send in parallel as many jobs as there are replicons in your data set.
The queue (or partition in SLURM terminology) with process.queue parameter (here common,dedicated)
and some options specific to your cluster management systems with process.clusterOptions parameter
cluster { executor { name = 'slurm' queueSize = 500 } process{ executor = 'slurm' queue= 'common,dedicated' clusterOptions = '--qos=fast' $integron_finder{ cpu=params.cpu } } }
To run the parallel version on a cluster, for instance on a cluster managed by slurm, I can launch the main nextflow process in one slot. The parallelization and the submission on the other slots is made by nextflow itself. Below a command line to run parallel_integron_finder and use 2 cpus per integron_finder task, each integron_finder task can be executed on a different machine, each integron_finder task claim 2 cores (cpus in nextflow terminology) to speed up the attC sites or integrase search:
sbatch --qos fast -p common nextflow run parallel_integron_finder.nf -profile cluster --replicons all_coli.fst --cpu 2 --local-max --gbk --circ
The results will be the same as described in local execution.
apptainer (formely singularity) profiles
If you use the integron_finder image with the apptainer container executor, use the profile standard_apptainer. With the command line below nextflow will download parallel_integron_finder from github and download the integron_finder image from the docker hub and convert it to apptainer on the fly so you haven’t to install anything except nextflow and apptainer.
nextflow run gem-pasteur/Integron_Finder -profile standard_apptainer --replicons all_coli.fst --circ
You can also use the integron_finder apptainer image on a cluster, for this use the profile cluster_apptainer.
sbatch --qos fast -p common nextflow run gem-pasteur/Integron_Finder:2.0 -profile cluster_apptainer --replicons all_coli.fst --cpu 2 --local-max --gbk --circ
In the case of your cluster cannot reach the world wide web. you have to download the apptainer image
apptainer pull --name Integron_Finder docker pull gempasteur/integron_finder:<tag>
the move the image on your cluster modify the nextflow.config to point on the location of the image, and adapt the cluster options (executor, queue, …) to your architecture
cluster_apptainer {
executor {
name = 'slurm'
queueSize = 500
}
process {
container = /path/to/integron_finder/image
queue = 'common,dedicated'
clusterOptions = '--qos=fast'
withName: integron_finder {
cpus = params.cpu
}
}
singularity {
enabled = true
runOptions = '-B /pasteur'
autoMounts = false
}
}
}
then run it
sbatch --qos fast -p common nextflow run ./parallel_integron_finder.nf -profile cluster_singualrity --replicons all_coli.fst --cpu 2 --local-max --gbk --circ
If you want to have more details about the jobs execution you can add some options to generate report:
Execution report
To enable the creation of this report add the -with-report command line option when
launching the pipeline execution. For example:
nextflow run ./parallel_integron_finder.nf -profile standard -with-report [file name] --replicons
It creates an HTML execution report: a single document which includes many useful metrics about a workflow execution. For further details see https://www.nextflow.io/docs/latest/tracing.html#execution-report
Trace report
In order to create the execution trace file add the -with-trace command line option when launching the pipeline
execution. For example:
nextflow run ./parallel_integron_finder.nf -profile standard -with-trace --replicons
It creates an HTML timeline for all processes executed in your pipeline. For further details see https://www.nextflow.io/docs/latest/tracing.html#timeline-report
Timeline report
To enable the creation of the timeline report add the -with-timeline
command line option when launching the pipeline execution. For example:
nextflow run ./parallel_integron_finder.nf -profile standard -with-timeline [file name] --replicons ...
It creates an execution tracing file that contains some useful information about each process executed in your pipeline script, including: submission time, start time, completion time, cpu and memory used. For further details see https://www.nextflow.io/docs/latest/tracing.html#trace-report
For integron diggers
Many options are set to prevent false positives. However, one may want higher sensitivity at the expense of having potentially false positives. Ultimately, only experimental experiments will tell whether a given attC sites or integrase is functional.
Also, note that because of how local_max works (ie. around already detected elements), true attC sites
may be found thanks to false attC sites, because false attC sites may trigger local_max around them.
Hence, one may want to use very relaxed parameters first with the --keep-tmp flag to rerun the analysis on
the same data while restrincting the parameters.
Clustering of elements
attC sites are clustered together if they are on the same strand and if they
are less than 4 kb apart (-dt 4000 by default). To cluster an array of attC sites and an integron
integrase, they also must be less than 4 kb apart. This value has been
empirically estimated and is consistent with previous observations showing that
biggest gene cassettes are about 2 kb long. This value of 4 kb can be modified
though:
integron_finder mysequences.fst --distance-thresh 10000
or, equivalently:
integron_finder mysequences.fst -dt 10000
This sets the threshold for clustering to 10 kb.
Note
The option --outdir allows you to chose the location of the Results folder (Results_Integron_Finder_mysequences).
If this folder already exists, IntegronFinder will not re-run analyses already done, except functional annotation.
It allows you to re-run rapidly IntegronFinder with a different --distance-thresh value.
Functional annotation needs to re-run each time because depending on the aggregation parameters,
the proteins associated with an integron might change.
Integrase
We use two HMM profiles for the detection of the integron integrase. One for tyrosine recombinase and one for a specific part of the integron integrase. To be specific we use the intersection of both hits, but one might want to use the union of both hits (and sees whether it exists cluster of attC sites nearby non integron-integrase…). To do so, use:
integron_finder mysequences.fst --union-integrases
attC evalue
The default evalue is 1. Sometimes, degenerated attC sites can have a evalue above 1 and one may want to increase this value to have a better sensitivity.
integron_finder mysequences.fst --evalue-attc 5
Here is a plot of how the sensitivity and false positive rate evolve as a function of the evalue:
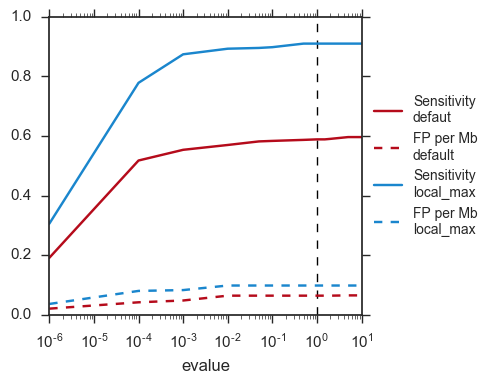
Note
If one wants to have maximum sensitivity, use a high evalue (max is 10), and then integron_finder can be run again on the same data with a lower evalue. It won’t work the other way around (starting with low evalue), as attC sites are not searched again.
attC size
By default, attC sites’ size ranges from 40 to 200bp. This can be changed with the --min-attc-size or --max-attc-size parameters:
integron_finder mysequences.fst --min-attc-size 50 --max-attc-size 100
Palindromes
attC sites are more or less palindromic sequences, and sometimes, a single attC site can be detected on the 2 strands. By default, the one with the highest evalue is discarded, but you can choose to keep them with the following option:
integron_finder mysequences.fst --keep-palindromes
attC alignements
One can get the alignements of attC sites in the temporary files (use --keep-tmp)
to have them. Under Results_Integron_Finder_mysequences/tmp_repliconA/repliconA_attc.res
one can find alignements of attC sites from repliconA, in Stokholm format, where R and L core regions
are aligned with each others:
# STOCKHOLM 1.0
#=GF AU Infernal 1.1.2
ACBA.0917.00019.0001/315102-315161 GUCUAACAAUUC---GUUCAAGCcgacgccgcu.................................................ucgcggcgcgGCUUAACUCAAGC----GUUAGAU
#=GR ACBA.0917.00019.0001/315102-315161 PP ************...******************.................................................***********************....*******
ACBA.0917.00019.0001/313260-313368 ACCUAACAAUUC---GUUCAAGCcgagaucgcuucgcggccgcggaguuguucggaaaaauugucacaacgccgcggccgcaaagcgcuccgGCUUAACUCAGGC----GUUGGGC
#=GR ACBA.0917.00019.0001/313260-313368 PP ************...******************************************************************************************....*******
ACBA.0917.00019.0001/313837-313906 GCCCAACAUGGC---GCUCAAGCcgaccggccagcccu.......................................gcgggcuguccgucgGCUUAGCUAGGGC----GUUAGAG
#=GR ACBA.0917.00019.0001/313837-313906 PP ************...***********************.......................................****************************....*******
#=GC SS_cons <<<<<<<--------<<<-<<<<.....................................................................>>>>>>>---------->>>>>>>
#=GC RF [Rsec=]========[=Lsec=].....................................................................[Lprim]==========[Rprim]
//
Which you can manipulate easily with esl-alimanip tools provided by infernal (the following examples should work if your cmsearch is in your PATH).
You can convert the same alignement in dna alphabet (cmsearch use RNA alphabet):
$ esl-alimanip --dna Results_Integron_Finder_mysequences/tmp_ACBA.0917.00019.0001/ACBA.0917.00019.0001_attc.res
# STOCKHOLM 1.0
#=GF AU Infernal 1.1.2
ACBA.0917.00019.0001/315102-315161 GTCTAACAATTC---GTTCAAGCCGACGCCGCT-------------------------------------------------TCGCGGCGCGGCTTAACTCAAGC----GTTAGAT
#=GR ACBA.0917.00019.0001/315102-315161 PP ************...******************.................................................***********************....*******
ACBA.0917.00019.0001/313260-313368 ACCTAACAATTC---GTTCAAGCCGAGATCGCTTCGCGGCCGCGGAGTTGTTCGGAAAAATTGTCACAACGCCGCGGCCGCAAAGCGCTCCGGCTTAACTCAGGC----GTTGGGC
#=GR ACBA.0917.00019.0001/313260-313368 PP ************...******************************************************************************************....*******
ACBA.0917.00019.0001/313837-313906 GCCCAACATGGC---GCTCAAGCCGACCGGCCAGCCCT---------------------------------------GCGGGCTGTCCGTCGGCTTAGCTAGGGC----GTTAGAG
#=GR ACBA.0917.00019.0001/313837-313906 PP ************...***********************.......................................****************************....*******
#=GC SS_cons <<<<<<<--------<<<-<<<<.....................................................................>>>>>>>---------->>>>>>>
#=GC RF [Rsec=]========[=Lsec=].....................................................................[Lprim]==========[Rprim]
//
You can also convert it to fasta format:
$ esl-alimanip --dna --outformat afa Results_Integron_Finder_mysequences/tmp_ACBA.0917.00019.0001/ACBA.0917.00019.0001_attc.res
>ACBA.0917.00019.0001/315102-315161
GTCTAACAATTC---GTTCAAGCCGACGCCGCT---------------------------
----------------------TCGCGGCGCGGCTTAACTCAAGC----GTTAGAT
>ACBA.0917.00019.0001/313260-313368
ACCTAACAATTC---GTTCAAGCCGAGATCGCTTCGCGGCCGCGGAGTTGTTCGGAAAAA
TTGTCACAACGCCGCGGCCGCAAAGCGCTCCGGCTTAACTCAGGC----GTTGGGC
>ACBA.0917.00019.0001/313837-313906
GCCCAACATGGC---GCTCAAGCCGACCGGCCAGCCCT----------------------
-----------------GCGGGCTGTCCGTCGGCTTAGCTAGGGC----GTTAGAG
The possible outformat are:
stockholm
pfam
a2m
psiblast
afa
